
Eight-year-old Philip Hopkins likes to joke that he’s “FDA approved” now that he’s in the second phase of a research trial for a recently approved cystic fibrosis treatment drug.
Philip was diagnosed with cystic fibrosis when he was just six weeks old, and he’s had the strong support of his dedicated family and a specialized team from our Cystic Fibrosis Center there for him every step of the way: “We love his CF team,” Philip’s mom Catherine said. “His doctor and his respiratory therapist are like family. They’ve known him since birth and we know we can call his team for anything. They’re always there to answer our questions and help with what he will need.”
.jpg)
CHoR has the only Cystic Fibrosis Foundation-accredited CF care center in Central Virginia and our program offers a multidisciplinary team of physicians, nurses and other specialists who provide comprehensive treatment for the specific issues associated with this lifelong condition. It is one of approximately 120 centers in the US accredited for providing expert care and specialized disease management.
Cystic fibrosis is a disorder that mainly affects functioning of the lungs and digestive system. It occurs when a child inherits a copy of a specific gene from each parent. CF causes cells in the lining of the lungs, digestive track and certain other organs to clog with mucus. When the mucus in the lungs is too thick, it cannot effectively do its job trapping and removing germs, which leads to a chronic respiratory tract infection that over time can lead to severe, non-reversible lung damage, limited breathing ability and even respiratory failure. In the digestive tract, the abnormal mucus blocks the pancreas from secreting digestive proteins and as a result many of the fats, vitamins and nutrients important for maintaining weight and fighting lung infections aren’t properly absorbed into the body.
A partnership for a healthier life
As an accredited center, CHoR is a recommended site for diagnostic testing for cystic fibrosis. Individuals with CF have a higher concentration of salt in their sweat and after newborn screening and other testing at CHoR, Philip underwent a sweat chloride test that confirmed he had cystic fibrosis.

Though Philip is the third of four children, he is the only one of his siblings with cystic fibrosis and since his diagnosis he and his family have worked closely with a specialized team led by pediatric pulmonologist and pediatric CF program director Dr. Joel Schmidt. “We work in partnership with our patients and their families to help make sure that their treatment plan and comprehensive CF care reflect their personal needs and goals so they can live longer and healthier lives,” said Jim Norton, a respiratory therapist who specializes in CF care and has worked closely with Philip through the years. These and other program goals align with the Cystic Fibrosis Foundation’s mission to promote individualized treatment and access to high-quality, specialized care to provide all people with the disease the opportunity to lead full, productive lives.
Both the daily and long-term goals of Philip’s treatment plan focus on ensuring that he and his parents have the care and support needed to maintain and maximize his lung function and nutritional status. “If his nutritional status declines, it can and likely will adversely affect his lung function and vice versa,” explained Jim.
During checkups Philip undergoes pulmonary function testing where the amount of air his lungs can hold and the rate at which air flows through his lungs are measured to create reports that guide targeted treatment decisions. Blood work, throat cultures and chest x-rays help the team further monitor his infection status and health. He also meets with Jim for guidance related to airway clearance issues and with the team’s dietitian for nutritional monitoring.
Other team specialists include a CF nurse specialist and social worker who work with Philip’s parents to ensure he has access to the medical devices and medications he needs and a psychologist who provides support managing the social and psychological challenges of living with a medical condition that requires a high level of daily care. “It’s very important to monitor our CF patients and their parents or caregivers for the development of depression or anxiety. We want our families to remain motivated to perform all the care their children need to maintain their health. We don’t want our patients or families to feel overwhelmed with the level of care required to remain healthy,” said Jim.
Daily treatments 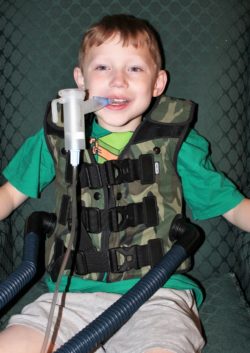
To keep his airways clear, Philip spends hours every day doing airway clearance therapy treatments to compensate for his impaired ability to clear his bronchial passages of infection. Two to three times a day, for about 30 minutes at a time, he wears a vest that vibrates to help move mucus out of his lungs so it doesn’t build up and exacerbate infections. While wearing the vest, he breathes in a highly concentrated salt water solution using a nebulizer which further assists clearing airways. The increased salt level in the aerosol mist the nebulizer releases attracts water into the airways that thins the mucus and improves the ability to cough up airway secretions.
To help his digestive system function well, Philip takes several medications, including pancreatic enzymes (the digestive proteins that are normally produced by the pancreas) before he eats to help his body better absorb calories and nutrients. He also takes a series of daily pills that help keep the bacteria in his digestive system healthy, help the enzymes work more efficiently, and minimize acid reflux which can be aggravated by the frequent coughing experienced by individuals with CF.
Support for eating issues
Throughout his life, Philip and his family have worked closely with the CF team’s dietitian on a specialized diet that incorporates specific foods to support weight gain and growth. “Since he doesn’t absorb fat well, it’s important for him to eat high-calorie, high-fat meals to maintain a healthy weight,” said Catherine.
In recent years, however, eating issues have become a complicating factor. Philip tends to eat smaller amounts which Catherine said is likely related to the physical effects of cystic fibrosis. He feels full quickly, possibly related to his slow-moving mucus, and at times experiences stomachaches due to digestion issues so pain is something he associates with eating. “Some kids with CF just don’t eat well, and he’s one of those kids,” she said.
This past May, Philip began treatment at our Children’s Feeding Program to help him develop eating patterns that can better support his nutritional needs. He comes for hour-long treatment sessions every other week with a speech therapist and psychologist from this program. “The goal is to get him to eat faster, before his brain tells him he’s full, and to get the maximum caloric content in small amount of food,” explained Catherine. “They’re also working on encouraging him to eat a larger variety of foods, more high-fat foods and more food at one sitting.”
Research & results
As a Cystic Fibrosis Foundation-accredited facility and member of the Cystic Fibrosis Therapeutics Development Network, which is the world’s largest network for CF clinical trials, our CF Center solicits patient participation in a wide range of CF clinical trials. “Our CF physicians and clinical research coordinators work to participate in CF related research trials to provide opportunities for our patients to assist in the development of new treatments and optimize chances for healthy lives for our patients,” explained Jim. “These trials are extremely crucial to help us identify, develop and test medications, therapies and diagnostic tools that will allow the CHoR CF team to improve, extend and optimize the health of our CF patient population.”
The year he turned 6, Philip began participating in the first phase of a clinical trial studying the safety and effectiveness of a new CF drug for children ages 6 to 11. The medication is designed to correct the function of the defective protein made by the CF gene and it had previously received FDA approval for ages 12 and above. “It’s not a cure, but it’s as close as we’ve come,” Catherine said. “Ideally, CF won’t progress in kids who take it at this age – they’ll have thinner mucus and won’t lose lung function over time.”
Starting March 2016, Philip took two pills twice a day for six months and was closely monitored by our clinical research team. This initial phase was a double-blind study, meaning some subjects were given the actual drug and others were given a placebo. Philip’s family later learned he did receive the drug. The medication has since been approved by the FDA for the 6 to 11 age group and Philip is now in the second phase of the trial where he will take it for two years and continue to be monitored by the research team. One of the lung function tests he undergoes as part of this is also being evaluated as a more sensitive way to safely detect and monitor small airway disease.
Maintaining control
According to Jim, Philip and his parents are doing an excellent job maintaining his respiratory and nutritional status in an optimal way. “This is due in no small part to his mother who leaves absolutely no stone unturned when it comes to obtaining the resources he needs to maximize his cystic fibrosis care,” he said.
One of the things Catherine and his team have worked toward – and that Catherine has been extremely pleased see – is that despite what is required of him on a daily basis, cystic fibrosis is not what defines Philip or his childhood experience. “He doesn’t let it control him,” said Catherine.
Philip loves basketball, reading and playing the piano. Those who know him describe him as bright, active, outgoing and ready for life’s next adventure with his siblings. (They just need to work it around his treatments.) He hopes to open a restaurant someday called Green P’s, where he plans to serve healthy – but tasty – versions of pizza, mac and cheese, and other kid favorites, putting some of what he’s learned from his dietitian to use.
Medication, therapy and monitoring have always been part of Philip’s life, and likely always will be. “He’s been doing this since he was a baby,” said Catherine. “He doesn’t fuss. He takes his pills like a champ and is OK with blood draws and other tests, especially at his clinical trial appointments. He’s just a go-with-the-flow sweet kid.”
This attitude is just what Philip needs for the next big step in his medical journey – handling more of his care on his own as he matures – and this is something his parents and team are already planning for together. “My hopes are that he will be independent in his therapies and that it won’t be a burden to him,” said Catherine. “I’d like to think by following his therapy guidelines and taking care of himself, he will maintain good lung function and live a normal life.”
The “Meet our Calendar Kids” blog series highlights children featured in CHoR’s Tid*Bits calendar. Join our mailing list to receive future issues of the Tid*Bits calendar and newsletters. We hope you’ve enjoyed getting to know Philip, our featured patient for June 2017.
More about Philip – Fun facts:
For fun: Basketball and reading
When he grows up: Run a restaurant called Green P’s
School: Homeschooled